Цистична фиброза
Синоними у ширем смислу
Цистична фиброза, плућа
Енглески: муковисцидоза, цистична фиброза
Дефиниција цистичне фиброзе
Цистична фиброза је наследна болест. Насљеђивање се медицински назива аутосомно рецесивно. Цистична фиброза (цистична фиброза) не наслеђује се на полним хромозомима Кс и И, већ на аутосомном хромозому 7.
Прочитајте наш општи чланак о метаболичким поремећајима: Метаболички поремећаји - шта то значи?
Мутација је на такозваном ЦФТР гену. Рецесивно је значило да морају постојати две неисправне копије гена да би болест избила. Ако особа има здраву и мутирану локацију гена на одговарајућем хромозому 7, болест се не јавља.
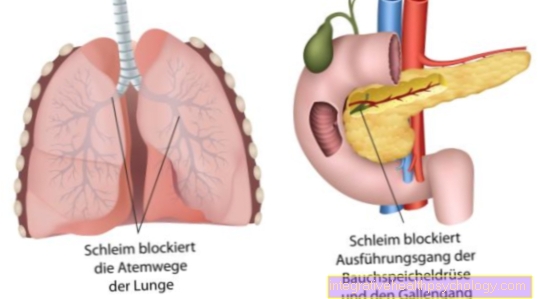
Резултат је патолошки генски производ. Тиме је кодиран Хлоридни канали су сломљени. Неисправни хлоридни канали доводе до стварања густе слузи у свим егзокриним жлездама.
Те егзокрине жлезде, тј. Жлезде које отпуштају спољашњу секрецију, укључују:
- гуштераче
- танког црева
- систем дисајних путева са плућима и бронхијалним системом
- билијарног тракта и
- такође Знојне жлезде
Резиме
Цистична фиброза је Насљедна болест. Насљеђује се на тај начин да је родно неовисан и само са њим два неисправна гена јавља. То је најчешће аутосомно рецесивно насљеђивање.
Последице су тешке творбе слузи свих егзокриних жлезда, попут жлезда плућа, панкреаса, као и знојних жлезда. Они су засновани на томе поремећен транспорт хлорида између унутрашње и спољашње ћелије (читај даље: Хлорид у крви). Мутирани ген је укључен Хромосом 7 и изазива велику разноликост захватања органа с одговарајућим ефектима на дисање, пробаву и репродукцију.
Нажалост, терапија може само ублажити симптоме, али не и лек. Тхе Очекивано трајање живота код пацијената са цистичном фиброзом релативно ниска.
Пошто се ради о рецесивној наследној болести, постоје људи који носе промењени ген, али не пате од саме болести. Такве особе се зову Носач функција или Проводницитј. носачи. Ови људи немају цистичну фиброзу јер је друга копија гена нетакнута, а болесна није довољно јака да превлада.
Међутим, она може пренијети ову неисправну копију гена свом потомству. Ако би модификовани ген већ био довољан да изазове болест, то би било такозвано доминантно наследство. Такво наслеђе може се наћи, на пример, у Цхореа хунтон. Више о овој болести можете сазнати у нашој теми Цхореа хунтон.
На око 1:2500 лаже оно Стопа болести код новорођенчади у Немачкој. Превозник се односи на све 25. у немачком становништву.
Корен

Цистична фиброза настаје мутацијом гена на хромозому 7. Овај хромозом је аутосомни хромозом, а не полни хромозом.
Свако има 44 аутосомална хромозома (две идентичне верзије сваког) и два полна хромозома. Ова мутација на хромозому 7 доводи до стварања неисправних хлоридних канала. Реапсорпција (поновна апсорпција) хлорида из жлездастих секрета није могућа јер рецептор, тачка прикључивања хлорида, није уграђен у канале жлезде.
Уместо тога, одложен је за рударство због неправилног изгледа и структуре. Природна размена хлорида кроз одређене хлоридне канале је поремећена. Ови такозвани канали сачињени су од протеина. Велики број протеина је кодиран на нашем ДНК. Због генетске оштећења хлоридних канала, долази до дехидриране и жилаве производње слузи из свих жлезда, које своју секрецију испуштају споља. Слуз затим дјелимично блокира канале или дисајне путеве у плућима.
Прочитајте и о овоме Мутација хромосома
Дијагноза цистичне фиброзе

Типични симптоми који почињу у дојеначкој доби су револуционарни у дијагнози цистичне фиброзе.
Ова сумња је појачана позитивном породичном историјом (болест оца / мајке или блиских рођака). Позитивна породична анамнеза значи да постоје или су већ били случајеви цистичне фиброзе у породици - на мајчиној или очевој страни.
Недостатак ензима панкреаса такође се може открити у столици. Било какве блокаде у дисајним путевима могу се открити рендгенском претрагом грудног коша.
Знојни тест, који мери садржај хлорида у зноју, такође помаже у дијагнози цистичне фиброзе. Ако је одређена вредност прекорачена, а примењују се и остали симптоми, дијагноза је релативно фиксна. Често родитељи сами примећују повећан садржај соли у зноју одојчета.
Нерођено дете се такође може тестирати на ову наследну болест. Коришћењем пункције амнионске течности (Амниоцентеза) ћелије фетуса се уклањају и испитују на мутирани ген.
Прочитајте више о теми: Рентгенски преглед детета
Терапија цистичне фиброзе
Свако погођен цистичном фиброзом добиће савет у једном Цистична фиброза - амбулантно одељење или савет од Људски генетичар (Препоручује се специјалиста за наследне болести). Ово може помоћи да повећате квалитет живота или, ако желите да имате децу, израчунајте вероватноћу болесног детета. Под условом да су родитељи плодни и плодни.
У супротном, лечење је симптоматско, јер узрок, неисправни ген не може бити елиминисан.
Неизлечива болест
Цистична фиброза (цистична фиброза) је и данас неизлечива болест.
У случају цистичне фиброзе важно је имати довољне количине кухињске солиНатријум-хлорид, НаЦл). Наравно је усмерена муколиза. Муколиза је растварање слузи, посебно у плућима, како би се олакшало дисање.
Лијекови и инхалације могу ублажити тегобе. Ако се функција плућа примјетно погорша, може се дати кисеоник.
Интензивном физиотерапијом (физиотерапија), на пример масажом тапкањем и вежбама дисања, лече се и промене плућа изазване цистичном фиброзом.
Често се болест заврши потребном трансплантацијом плућа. Међутим, листе чекања су дуге.
Перорална примена ензима панкреаса и витамина растворљивих у мастима такође је део терапије. Задатак гуштераче се стога мора подржати, или боље заменити. У мастима су растворљиви у мастима А, Д, Е и К. Они се морају давати директно у крв јер се не могу апсорбовати из хране због недостатка пробавних ензима.
У исхрани би такође требало бити пуно калорија, јер се из хране може добити само делић њих.
Да би се избегли додатни фактори ризика од компликација, попут грипа или упале плућа, дете треба вакцинисати. Препоручују се следећа вакцинације:
- оспице
- Пнеумококи
- грип
Прочитајте више о теми: Суперинфекција
Наравно, за те мере је потребна консултација са лекаром, са којим би се требало разговарати о ризицима.
Данас се велика нада за терапију цистичне фиброзе полаже у генетичка истраживања. Покушава се увести недостајући генетски податак у геном човека. Тражимо векторе који би могли да савладају овај задатак. Вектори могу, на пример, бити бактеријска или вирусна ДНК која успева да угради здраву фреквенцију у наш генетски састав.
Тренутно се тестира терапијски приступ код нерођених пацијената. Код мишева, мишји ембриони су већ успели да уводе здрав ген, који је садржао исправан генски низ, амниоцентезом (инокулацијом амнионске течности). Тако је добијен здрав ЦФТР ген код ових мишева. Амниоцентеза је пробијање и уклањање дечијих ћелија из амнионске течности, што се врши кроз мајчин трбушни зид.
Међутим, у Немачкој је овај облик интраутерине (= у матерници = у матерници) забрањен "терапија".
профилакса
А превентивна мера у том смислу не постоји, јер је наследна болест.
Међутим, може се посетити и хумани генетски саветодавни центар (који се обично налази у универзитетским болницама). Овде се израчунава колики би ризик био да се болест пренесе на децу.
Овај савет је увек користан ако постоји породична историја цистичне фиброзе.
Такође један Пренатална дијагностика вреди тежити. Овде пре рођења (тј. Пренатално) а Преглед амнионске течности (Амниоцентеза) спроведена. Феталне ћелије (ћелије детета) узимају се из амнионске течности и ДНК се испитује на мутирани ген.
Прогноза цистичне фиброзе
Нажалост, просечан животни век пацијената са цистичном фиброзом је свега 32-37 година. Данас се животни век новорођенчади рођених са овим стањем процењује на око 45-50 година.
Прогноза јако зависи од терапије и од тога да ли се је придржава.
Сам пацијент и његова мотивација играју важну улогу.








.jpg)