хемофилија
Синоними
Хемофилија, наследни поремећај крварења, недостатак фактора згрушавања крви,
Недостатак фактора ВИИИ, дефицит фактора ИКС, хемофилија
дефиниција
Хемофилија је наследна болест система згрушавања крви:
Болесници који су погођени имају оштећење згрушавања крви, што се манифестује чињеницом да крварење настаје са најмањом траумом и крварење је тешко зауставити.
Фактори коагулације се не могу активирати, тако да редовни ток каскаде коагулације није могућ.
Хемофилија је урођена болест која се јавља у два облика,
Хемофилија А и Б, може бити присутна.
Хемофилија А се јавља чешће (у 85% случајева) од облика Б (приближно 15%) и озбиљнија је од два облика.
Активност комплекса фактора ВИИИ је ограничена код хемофилије А, јер је стварање фактора ВИИИ-Ц у ћелијама јетре смањено.
Фактор ВИИИ-Ц једна је од две подјединице комплекса фактора ВИИИ:Виллебранд-Фактор и фактор ВИИИ-Ц формирају овај комплекс који потиче коагулацију у каскади коагулације. Фон Вилебрандов фактор штити фактор ВИИИ-Ц од препадања.
Количина функционалног фактора ВИИИ комплекса се смањује у облику А.
У хемофилија Б. синтеза фактора коагулације ИКС се одвија у мањем обиму, тако да његов ефекат у процесу коагулације није присутан и хемостаза је ослабљена.
Појава / епидемиологија
Појава хемофилије у популацији је за хемофилију А. 1 на 5.000 и за облик Б. 1:15.000.
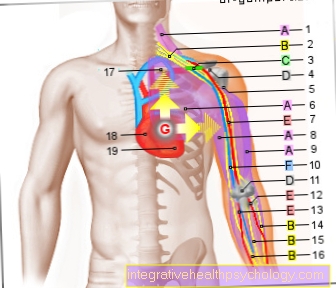
Чињеница да се мушкарци посебно разболеју због родних питања
Кс-везани облик насљеђивања оштећења коагулације (види узрок / развој).
Основе
Коагулација крви је фино подешен систем: Различите компоненте крви, такозвани фактори коагулације, активирају се један за другим на каскадни начин како би се омогућила хемостаза у случају повреде.
Систем коагулације састоји се од примарне и секундарне хемостазе, која се још назива и плазматска коагулација.
Ако је зид жиле повређен, захваћени жила сужава се у првом кораку (= примарна коагулација). Тада је проп (=Тромбоцити тромбоцита) на повређеном делу који је покривен тромбоцитима (= Тромбоцити) формира се ради постизања почетног печата посуде.
Формирање тромба активира плазмасту коагулацију:
Почиње каскада коагулације и интеракција различитих фактора коагулације ствара стабилно затварање ране или васкула.
Узрок и порекло
Тхе хемофилија Оба облика хемофилије су наслеђена као Кс-везана рецесивна особина.
У такозваном Кс-везаном начину насљеђивања хемофилије, генетска информација за генски производ налази се на сполном хромозому Кс, који се налази у двоструком броју код жена и у једном броју код мушкараца.
Постоје два гена за сваку генетску информацију, такозвани алели. Ако постоји рецесивно наследство, оба алела морају бити под утицајем мутације да би проузроковала болест: Рецесивна значи да патолошки измењене генетске информације потичу од здравих, тј. не мења се, информације се потискују и не доводе до болести. Рецесивна насљедна болест настаје само ако оба алела носе погрешне генетске информације.
Је само а Израз гена се променио, хајде Не до хемофилија.
Инфо: наслеђивање
Ако се промени само једна експресија гена, неће бити болести.
Пошто мушкарци имају само један Кс хромозом у пару сполних хромозома и зато су информације о генским производима на њиховом Кс хромозому доступне само у једноставном облику, разболеће се ако је само један алел неисправан.
Жене, с друге стране, које имају два Кс хромозома као полне хромозоме, постају болесне само када оба хромозома имају неисправне гене за генски производ.
Код мушкараца који пате од хемофилије, алел који садржи наследне информације за фактор коагулације ВИИИ-Ц (хемофилија А) или фактор коагулације ИКС (хемофилија Б) доступан је само у мало измењеном облику;
Жене морају да носе ову генетску промену на оба алела да би биле болесне.
Ако жене имају неисправан и неизмењен алел, називају се носиоцима наследне болести Кс-везе, као што је хемофилија.
Код хемофилије, неисправне генетске информације имају за последицу смањење активности погођеног фактора коагулације или смањење производње фактора коагулације.
Кс хромозом је носилац генетског материјала за факторе коагулације секундарне хемостазе.
Приближно 70% мутација у генетском материјалу које узрокују хемофилију преноси се у породицама, а 30% болести потиче од спонтаних промена (= спонтаних мутација) у генетским информацијама.
дијагноза
Након што се распитате о здравственој историји пацијента и обавите темељит физички преглед, следе додатни кораци у дијагностици хемофилије:
У 2/3 случајева има случајева хемофилије у породици, због чега се мора питати породично групирање болести ако се пацијент покаже лекару са симптомима сумњивим на хемофилију.
Пацијенти пријављују модрице као посљедицу најмањих повреда.
Један од њих разликује Тежина хемофилије у благи, умерени или тешки облик болести.
Преглед узорка крви даје следеће резултате типичне за хемофилију:
Време крварења је нормално (= примарна коагулација је нетакнута), али функција плазматске коагулације је смањена, због чега је такозвано време ПТТ дуже.
ПТТ означава време делимичног тромбопластина. То се мери лабораторијским тестовима како би се проверила функција фактора И, ИИ, В, ВИИИ до КСИИ као и КСИВ и КСВ.
Будући да један фактор у каскади коагулације недостаје у хемофилији, он се не може одвијати на оптималан начин, што резултира продуженим временом коагулације.
Да би се могла разликовати хемофилија А и Б, крв болесника мора се испитати на фактори ВИИИ и ИКС, а одсуство два фактора одређује врсту хемофилије.
Диференцијална дијагноза
Хемофилија се мора разликовати од осталих поремећаја система коагулације, посебно се помиње вон Виллебранд-Јургенс синдром.
Фактор вон Виллебранд (вВФ) делује заједно са фактором ВИИИ-Ц им
Комплекс фактора ВИИИ и изазива инхибицију преурањене деградације фактора ВИИИ, фактор такође поспјешује коагулацију посредством адхезије крвних плочица на повређеном васкуларном месту. Стога је вВФ важан део и примарне и секундарне коагулације.
Ако је присутан синдром Вилебранд-Јиргенс, тада настаје
вВФ у ћелијама стијенке посуде одвијају се само у малој мери или на погрешан начин. Смањена стопа образовања или неадекватна функција
вон Виллебрандов фактор је узрокован мутацијама гена за формирање фактора. Насљеђивање синдрома или мутације узрочних гена насљеђује се на аутосомно доминантан начин: генетска информација за вВФ је на хромосому број 12, што није полни хромозом. У доминантном начину насљеђивања, болесни алел сузбија ефекат другог, здравог алела, тако да болест настаје мутацијом гена и изазива клиничке симптоме.
Пацијенти обично пате од крварења из коже и слузокоже, а мање од спонтаних крварења као код хемофилија.
Болест се често примети тек када после операције постоји дуготрајно крварење.
Време крварења пацијента после повреде се продужава, а вредности плазматске коагулације патолошки се мењају (продужено ПТТ).
Вон Виллебранд-Јургенс синдром узрокован је примјеном десмопресина или замјеном фактора ВИИИ и вВФ ради стабилизирања коагулације (детаљније објашњење видјети у одјељку „Терапија хемофилије“).
Који су симптоми хемофилије?
Симптоми у облицима хемофилије се не разликују:
- Хемофилија обично води само до клиничких симптома код мушкараца.
- Прекомерно крварење јавља се код хемофилије, што није у односу на претходну, углавном баналну несрећу (траума), време крварења је нормално, али обично долази до крварења које се не јавља код здравих људи.
- Разликује се између три облика хемофилије, који су дефинисани резидуалном активношћу погођених фактора коагулације:
1.тешка хемофилија (хемофилија) са мање од 1% заостале активности или дела функционалног фактора који је и даље присутан у којем долази до спонтаног крварења и крварења у зглобовима
2. Умерена хемофилија (крвна болест) са факторском активношћу која варира између
1 и 5% нормалне факторске активности и појава модрица (= хематома) након мање трауме
3. блага хемофилија (хемофилија) у којој још увек постоји 5-15% резидуалне активности и код којих пацијенти извештавају о симптомима као што су хематоми (модрице) након значајне трауме и постоперативног крварења.
Прочитајте више о теми: Који су узроци можданог крварења
- Жене које обично имају само измењене генетске информације о хемофилији обично не показују клиничке симптоме са преко 50% заостале активности.
- Пацијенти осећају крварење у велике зглобове попут тога колена-, Хип- или Зглоб рамена, при чему се говори о хемартхросис. Крварење покреће упалну реакцију са поправним процесима у зглобу, што може довести до укрућења зглоба.
- Такође може да искрвари у Мусцулатуре и мека ткива долазе. Крварење доводи до повећања притиска у мишићима или ткиву, јер је сада већа количина. Ово посебно може утицати на ваше руке и ноге Синдром одељка да води:
Повећани притисак изазива компресију крвних судова и живаца, тако да екстремитети нису довољно потрошени и велике површине ткива могу умрети. Одјељак синдром мора лијечити хирург што је прије могуће како би се спријечио губитак екстремитета. - Појављује се крварење у трбуху, што је за живот пацијента опасна ситуација.
- Необично дуготрајно крварење је могуће након операције.
Поред тога, могу бити дужи периоди са крвљу у урину. Овде једна прети Анемијајер пацијент стално губи крв, вероватно непримећено. - Посебно су опасни Церебрално крварење (= интракранијално крварење), што доводи до смрти код 10% људи са хемофилијом. Више о овој теми можете сазнати у нашој теми Церебрално крварење.
Како се изводи терапија за хемофилију?

Крварење би требало да се појави код болесних хемофилија мора се избегавати, због чега пацијент не Лекови које инхибирају згрушавање крви попут Ацетилсалицилна киселина (Аспирин®) и без интрамускуларних ињекција (= шприцеви у Мусцулатуре) редом.
Ако се догоди траума са крварењем, пажљива локална хемостаза је од великог значаја како би се спречило крварење у суседно ткиво.
Терапија лековима хемофилија састоји се у замјени фактора коагулације који тело не може произвести у довољним количинама.
Пацијенти са благим хемофилија по потреби примају препарате фактора коагулације, тј. ако је дошло до трауме са крварењем или ако се планира велика операција, што може резултирати обилним крварењем.
Замјена профилактичког фактора треба користити код пацијената са умјереном до тешком хемофилијом, тј. фактор који недостаје треба дати трајно, пре него што настаје крварење.
Ако је резидуална активност фактора ВИИИ већа од 15% или фактора ИКС већа од 20-25%, редовна терапија није неопходна; ови пацијенти добијају фактор коагулације који недостаје у случају спонтаног крварења или пре планиране хирургије.
Дуготрајна терапија, као и она пре операције, може бити у облику кућне само-лечења, у којој пацијент сам примењује фактор коагулације који недостаје.
За пацијенте са благом хемофилијом постоји алтернатива за терапију:
Активна супстанца Десмопрессин (нпр. Минирин®) доводи до дистрибуције
Фактор ВИИИ, који се чува у зидовима посуда.
Међутим, лек се може давати само неколико дана, пошто се након стимулације ослобађања фактора памћење у зидовима посуда исцрпи и мора се поново поновити.
У акутној терапији хемофилија постоје три опције за интервенцију:
- Пацијент прима активирани протромбин, супстанцу која потиче згрушавање крви тако да се крв заустави што је брже могуће.
- Друга терапијска опција је примена препарата фактора ВИИ. Овај фактор је на почетку каскаде коагулације и започиње свој редован ток.
- Трећа терапијска опција је примена животињског фактора ВИИИ-Ц како би се омогућило пацијенту да заустави крварење.
Компликације
Заменски поклон (=супституција) фактора коагулације могу довести до стварања антитела против управо тих фактора, тако да супституција у константној дози више нема терапијске ефекте, због чега се говори о стварању инхибитора.
У зависности од одређивања концентрације антитела у пацијентовој крви, могу се применити високе дозе фактора ВИИИ, чији је циљ повратити толеранцију на овај фактор и искључити стварање антитела. Овај поступак треба извести само у специјализованим центрима.
Постоји мали ризик од заразе хепатитисом или заразе вирусом ХИВ-а (=ХИВ) јер су фактори изведени из људских крви.
Други ризик супституционе терапије за хемофилију је појава алергијских реакција.